Beyond CD3: Next-gen T cell Engagers with Improved Specificity
This article refers to conventional T cell engaging bispecific antibodies. For a comprehensive comparison of approved T cell therapies refer to the following recent review manuscript by Baeuerle et al., https://doi.org/10.1084/jem.20251652.
A dire need for improved strategy of TCEs in treatment of solid tumors is apparent from the following timeline:
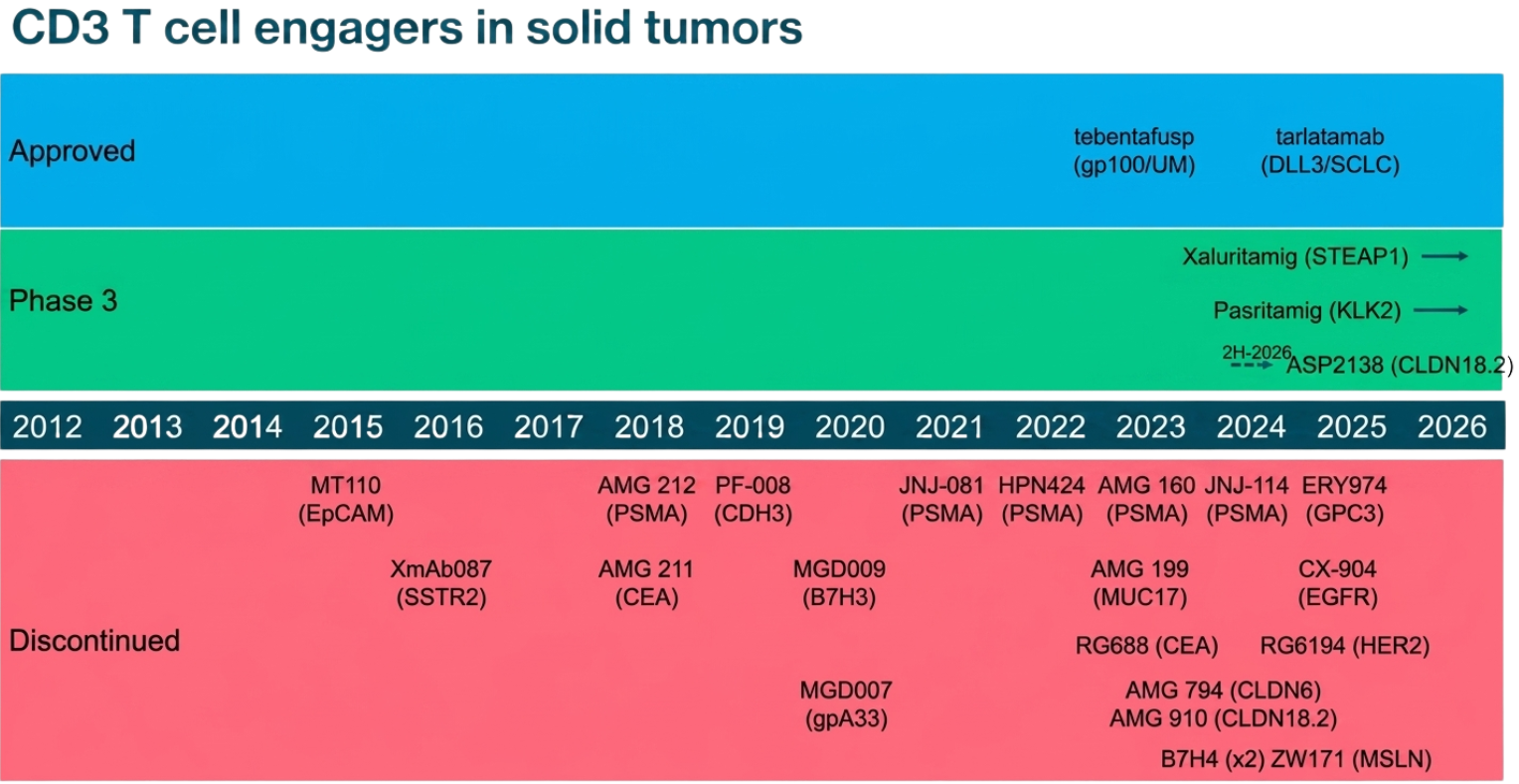
A timeline of T cell engagers (TCEs) in solid tumors, grouped by development status from 2012to today.
The main 3 improvement strategies for next gen TCEs:
Engaging Effector CD8+ cell subset instead of pan T cells
Stimulating Memory T cells instead of Naive T cells
Targeting TCR and not CD3
The harsh reality is that the approved T cell engaging therapeutics (TCEs) have thus far surpassed the expectations of many (onco)immunologists in their clinical efficacy, simply because the current therapeutics all provide a rather non-specific and non-physiological stimulation of the entire T cell population (pan T cell engagers). Nevertheless, the pitfalls of these engagers are becoming more evident with many cancer patients being refractory or building adaptive resistance. Furthermore, as T cell engagers are becoming increasingly interesting in autoimmune setting, the tolerance for suboptimal and inadequate responses is shrinking and our expectations and standards are growing with each new approved TCE.
TCEs represent a targeted immunotherapy approach that has reshaped the cancer treatment, similarly as immune checkpoint blockers (ICB) did, but operating via distinct mechanism of action (MOA). While ICB unleash the immuno-suppressive signals (‘the break’) preventing proper immune surveillance of tumor cells, TCE present a much more “synthetic” approach.
TCE are antibody-based constructs designed to transiently redirect T cells for target cell elimination. TCEs circumvent the need for an intrinsically generated anti-tumor immunity by redirecting T cells to form an immune synapse with target cells, inducing T cell activation, transient cytokine production, and ultimately tumor (i.e. target) cell lysis. Fundamental to the structure of all of the current TCEs are two distinct domains: one that binds a tumor cell surface antigen while the other engages CD3 on T cells.
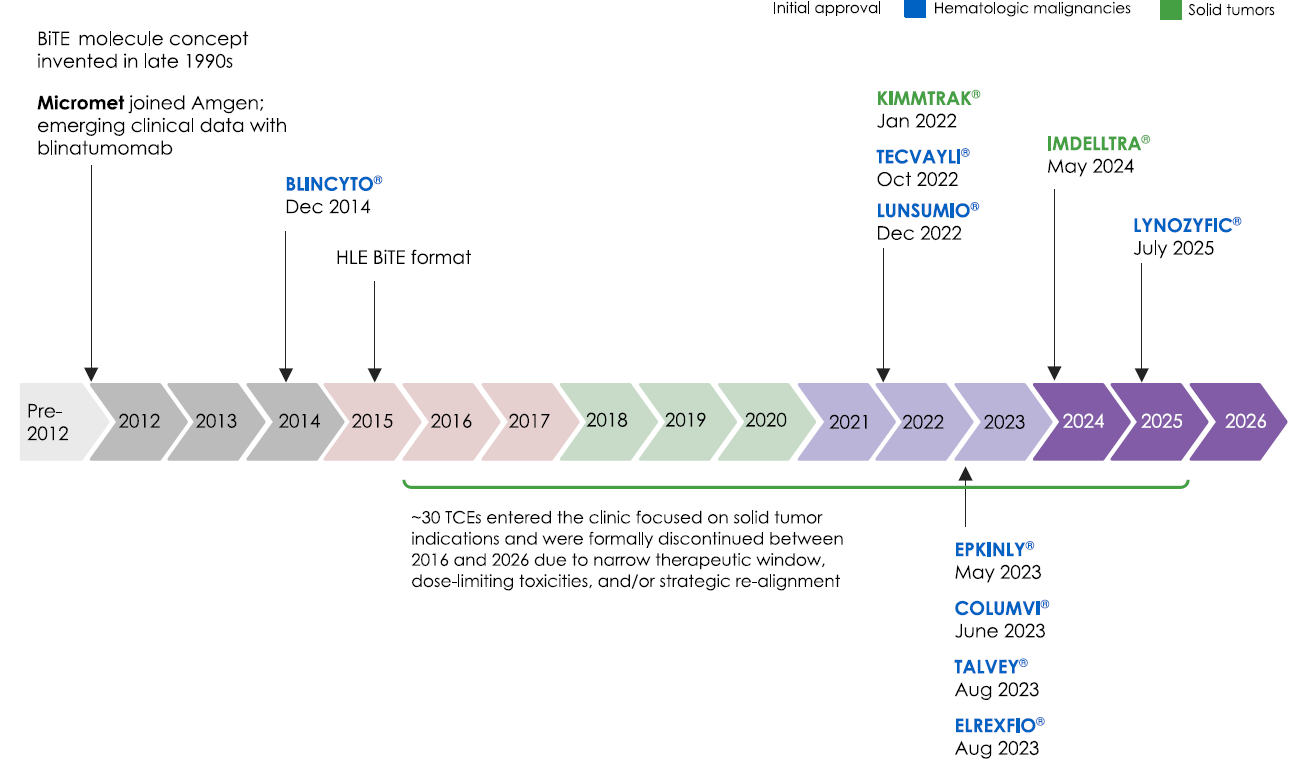
Over the past 12 years, 10 TCEs were approved by the US Food and Drug Administration, and an additional two by the European Medicines Agency. Nine TCEs treat hematologic malignancies, and three target solid tumors. Over 150 TCEs are being investigated in clinical trials, recently also in autoimmune diseases. (https://doi.org/10.1016/j.ccell.2026.03.011 )
Targeting T cells: CD8+ vs CD4+ subsets
By engaging CD3, more specifically CD3ε, TCE are binding to and recruiting the entire T cell population. This is one of the first pitfalls that directly prompts an immunologist with a question: “Why would one want to recruit all the T cells ? “ Namely, cytotoxic CD8+ T cells (CTLs) are highly potent effector cells to recognize and kill tumor cells expressing aberrant neoantigens, while the rest of the cells in the best case support their activity. More generally however, majority of T cells present inexperienced (i.e. naive) population, with CD4+ T cells serving to provide co-stimulatory signals and secrete cytokines, that can divert the immune responses. CD4+ T cells and cytokines are detrimental in dictating the therapeutic window of many TCE, by eliciting so called cytokine release storm (CRS). Furthermore, a subset of T cells called regulatory T cells actively inhibits anti-tumor immune responses. Yet, all the current TCE in the clinic are redirecting all the T cells by engaging CD3ε subunit.
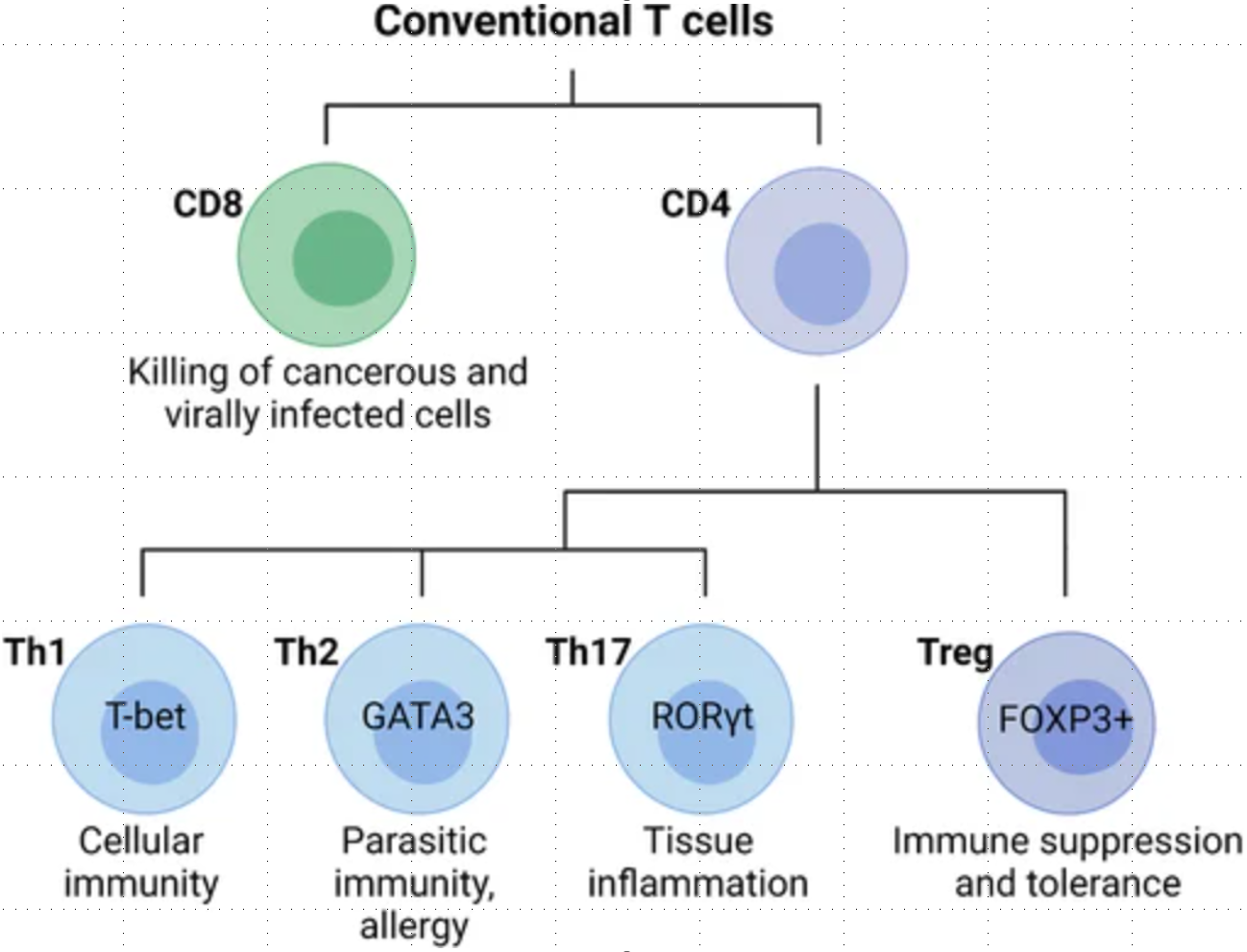
Conventional T cells are broadly grouped into CD8 and CD4 T cell subsets with diverse functions during an immune response. In general, CD8 cytotoxic T cells directly kill cancerous or infected cells, while CD4 T cells regulate the immune response to a particular antigen. CD4 T cells can be classified as T helper (Th) cells or regulatory T cells (Tregs). Differentiated CD4 T cell subsets are marked by the expression of the lineage-specific transcription factors T-bet (Th1), GATA3 (Th2), RORγt (Th17), and FOXP3 (Tregs), which are critical for their differentiation and function.Conventional T cells are broadly grouped into CD8 and CD4 T cell subsets with diverse functions during an immune response. In general, CD8 cytotoxic T cells directly kill cancerous or infected cells, while CD4 T cells regulate the immune response to a particular antigen. CD4 T cells can be classified as T helper (Th) cells or regulatory T cells (Tregs). Differentiated CD4 T cell subsets are marked by the expression of the lineage-specific transcription factors T-bet (Th1), GATA3 (Th2), RORγt (Th17), and FOXP3 (Tregs), which are critical for their differentiation and function.
Effector CD8+ T cells are critical for responses to TCE therapy
In line with previous studies investigating response patterns to ICB where absolute clonotype expansion correlates with clinical response, an effective TCE response by T cells is dependent on clonal expansion of CD8 effector cells.
Targeting Memory T cell populations to circumvent the need for balanced delivery of Signals 1-4 required by Naive T cells
In addition to clonal expansion or homing of pre-existing clones, the increased abundance of effector CD8+ T cells in TCE-treated patients might be the result of an induced phenotypic shift of naive T cells to effector states.
However, one important thing to note about the naive T cells is that these are high maintenance cells, requiring a combination of stimuli for proper conversion to effector state. Naive CD8+ T cells need to interact with licensed antigen presenting cells, with the help of CD4+ Th cells in order to acquire not only Signal 1 (TCRαβ), and Signal 2 (co-stimulatory signal CD28), but also Signal 3 (cytokines IL2, IFN-g, ….) and Signal 4 (proper nutrients). It takes a village.
Tumor cells typically do not express e.g. CD28 ligands (CD80/86), which compromises the activity of CD3 TCE. For this reason, many current approaches to improve TCEs are focused on integrating co-stimulatory signals (CD2, CD28, 4-1BB, ICOS agonistic arms - for comparison of different co-receptors see Sergeeva and Jin et al. PNAS 2025, https://doi.org/10.1073/pnas.2510829122) in a tri-specific antibody design.
Co-stimulation is meant to yield differentiated and superior CD8+ T effector-memory profile. A few recent examples:
anti-CD3 x CD28 x TAAs (e.g. XmAb808, JNJ-9401 and JNJ-1493 by Xencor & partners, ZW209 by Zymeworks, novel HLX390 by Shanghai Henlius Biotech, discovery of CD28 targeting single domain antibodies by Wuxi Biologics, FPE021 by Fapon Biopharma) or
anti-CD3 x CD2 x TAA (e.g. QL535 by QLSF Biotehrapeutics, affinity Tuned EVOLVE platform with the EVOLVE104 by EVOLVEImmune, GS24-B047 by Changchun GeneScience Pharmaceutical) in preclinical development are aiming to enhance and prolong anti-tumor response.
anti-CD3 x 4-1BB x TAA (e.g. BCG024 by Biocytogen)
In contrast, memory T cells can respond with less dependence on classic co-stimulation. In fact, effector CTLs rely on a different repertoire of co-stimulatory signals that may be provided by tumor cells (Dong B, Obermajer N, et al., JITC 2024 https://doi.org/10.1158/2326-6066.CIR-24-0061 ).
The ultimate idea of the next generation TCEs is that by redirecting memory T cells we would be able to circumvent the need to include co-stimulation.
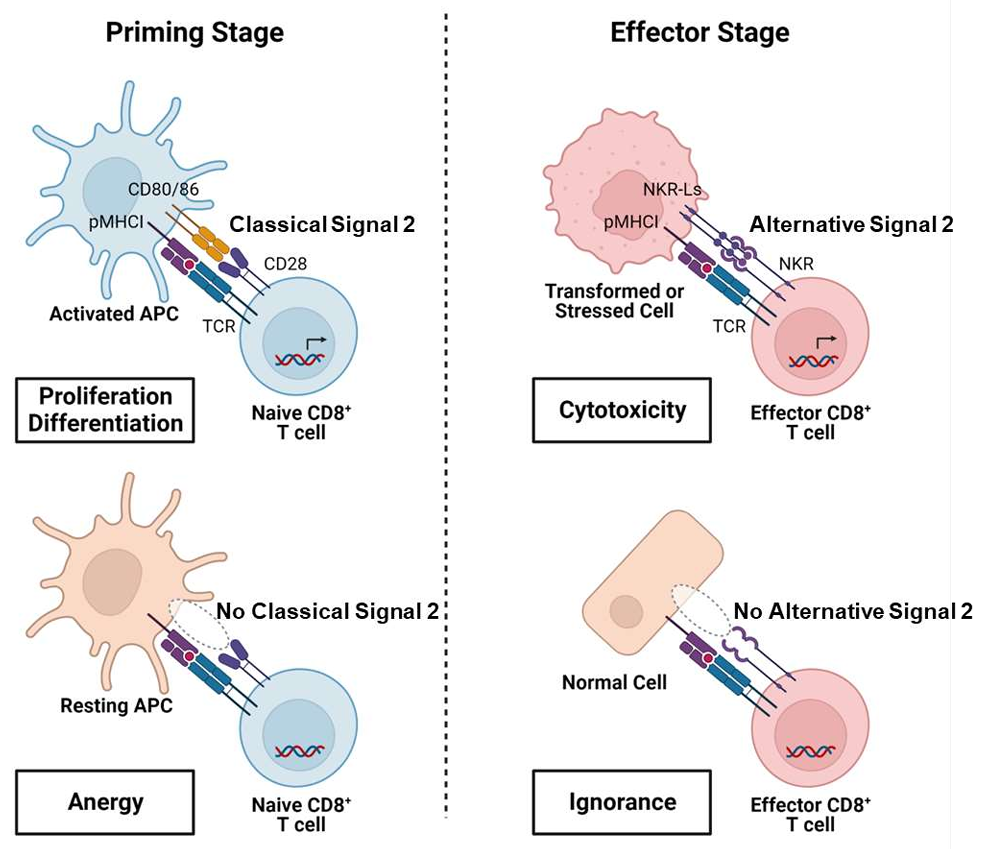
A model of NKR-mediated “alternative signal 2” supporting the specificity of CTL anti-cancer function. At the priming stage, CD28, by binding of APC-expressed CD28 ligands, provides classical signal 2 to costimulate the activation of naïve CD8+ T cells which are specific for antigens presented by activated APCs. In contrast, naïve CD8+ T cells specific for “self” antigens presented by resting APCs, are only partially activated in the absence of appropriate costimulation, which result in T-cell anergy. At the effector stage, effector CD8+ T cells become able to use NKRs to recognize NKR ligands expressed at higher levels on stressed cells, including infected, damaged or cancer cells, allowing them to receive “alternative signal 2” as a replacement for the missing CD28 costimulation, to support their CTL effector function. This allows CTLs specific for TAAs, which are shared between cancer and healthy cells, to ignore healthy cells, due to the lack of NKR-mediated co-stimulation, thus enhancing “self/non-self” discrimination.
Targeting T cells: CD3ε ≠ TCRαβ
This is probably one of the most instrumental misconceptions in immunology.
TCRαβ and CD3 form a structural and functional octameric complex, with the TCR mediating antigen-recognition and CD3 providing the signaling module necessary for T-cell activation. Nevertheless, targeting CD3ε is not equal to stimulating T cell receptor (TCR, specifically TCRαβ). Already early work conducted with CD3 mAb OKT-3 and TCRαβ mAb BMA031 indicated that TCRαβ stimulation by BMA031 was able to induce T-cell activation but did not lead to systemic adverse reactions after its administration to patients, contrarily to OKT-3 (see JCI 1992 publication: https://doi.org/10.1007/BF00918085 .

Administration of OKT3 or BMA031 is able to induce definite signs of T cell activation in patients. This activation is accompanied in the case of OKT3 (CD3ε) with clinical side effects, whereas in the case of BMA031 (TCRαβ) clinical symptoms do not occur. https://doi.org/10.1007/BF00918085
TCR-CD3 complex
The pioneering work by Drs. Mak and Davis in 1984 led to our current comprehension of the TCR complex as an eight-part receptor with intricate signaling pathways and functions. The tribute article to the groundbreaking achievement of TCR cloning has been published in 2024: https://doi.org/10.1038/s41423-024-01168-4 .
The TCR–CD3 complex is a multi-subunit protein complex. It is composed of an antigen-binding TCRαβ heterodimer (TCR) non-covalently associated with the signal transduction subunits; the CD3 heterodimers CD3εγ and CD3εδ as well as the CD3ζζ homodimers. The intracellular portions of TCR chains are too short to signal; therefore, TCRs rely on the CD3 complex to transduce intracellular signals upon activation.
The cytoplasmic tails of CD3 subunits contain activation motifs (ITAMs). Antigen binding to TCRαβ activates TCR-CD3 complex, leading to recruitment and activation of multiple downstream signaling molecules that have been reported over the past decades, including enzymes and adaptor proteins.
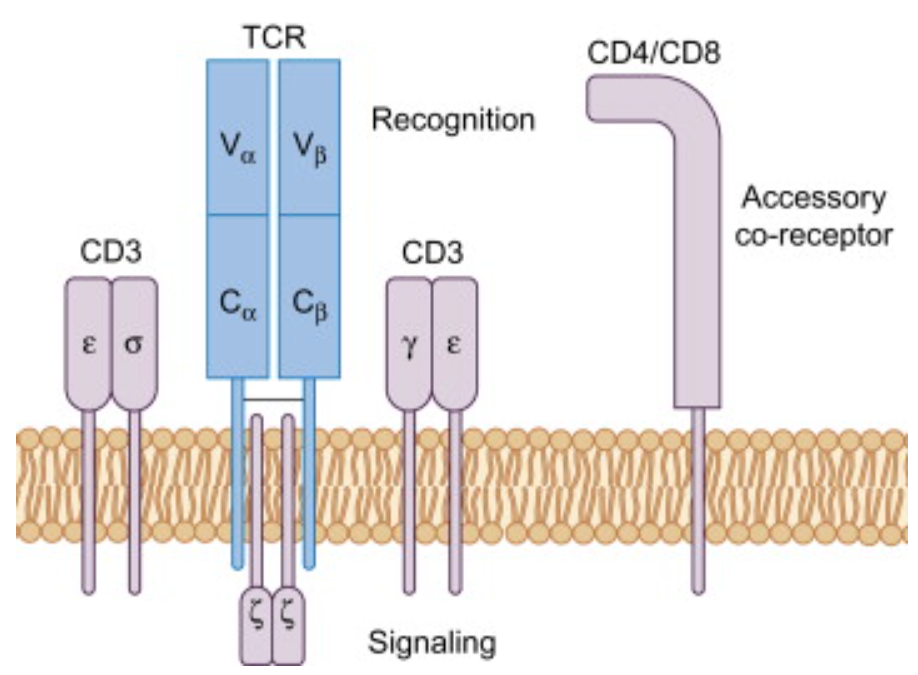
The T-cell receptor (TCR)–CD3 complex, expressed on T cells, determines the outcome of a T-cell response. It consists of the TCR-αβ heterodimer and the non-covalently associated signalling dimers of CD3εγ, CD3εδ and CD3ζζ. TCR-αβ binds specifically to a cognate peptide antigen bound to an MHC molecule, whereas the CD3 subunits transmit the signal into the cytosol to activate signalling events. Recruitment of proteins to specialized localizations is one mechanism to regulate activation and termination of signalling. In the last 25 years a large number of signalling molecules recruited to the TCR–CD3 complex upon antigen binding to TCR-αβ have been described. https://doi.org/10.1016/B978-0-12-420030-2.00004-4
Resistance Alert: CD3ε TCEs require TCR co-engagement to exert full activity and Tumors may exploit this vulnerability
Response to TCEs targeting CD3ε is wrongfully perceived sufficient to fully activate all the T cells and to be independent of tumor recognition and T cell state. Recently, Friedrich et al. provided new insights how T cells in cancer patients respond to TCEs. It turns out that T cell expansion and effector differentiation induced by CD3ε TCEs require additional TCR activation signals. See: https://doi.org/10.1016/j.ccell.2023.02.008
Because TCR co-stimulation amplifies overall T cell response to TCEs by functional recruitment and priming of naive T cell clones, loss of MHC class I may present a tumor-intrinsic mechanism of acquired resistance for CD3ε TCEs specifically.
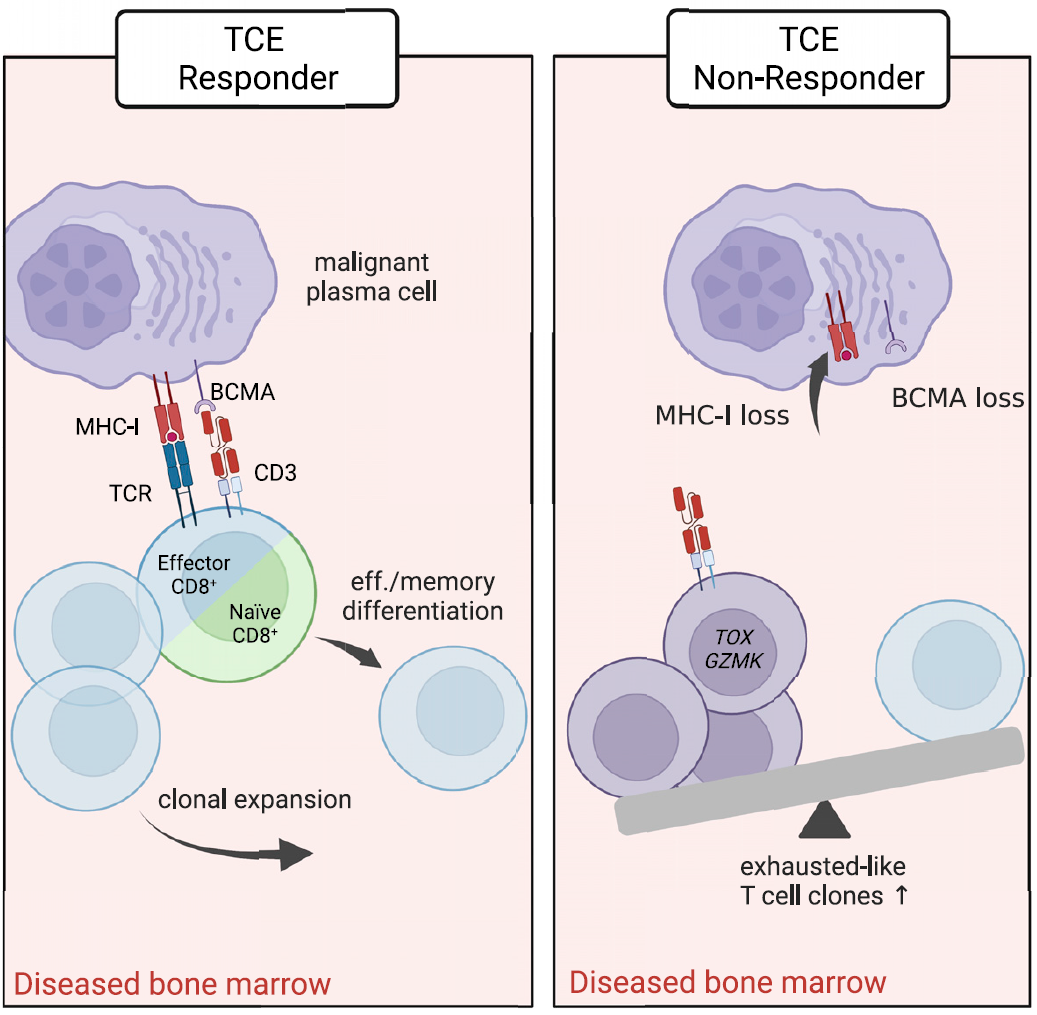
TCE-reactive T cells require additional TCR signal to differentiate upon TCE activation. The expansion of TCE-reactive TCRs is significantly reduced by blockade of TMHC class I:TCR signaling. https://doi.org/10.1016/j.ccell.2023.02.008
Next Gen Approaches Highlights
In contrast to the above mentioned improvements of TCEs, which aid as much as an oil exchange to a vehicle with a broken engine, the below examples do present substantial improvements into the right direction.
Activation of specific T cell subsets
T cell subset selectivity represents one step to improve the therapeutic index for TCEs by reducing the number of T cells activated and lowering CRS-associated toxicities. As the CD8+ T cells are primarily responsible for mediating cytotoxicity against (malignant) target cells and play a crucial role in anti-cancer immune responses, several examples aim to engage this subset of T cells.
Understanding T cell biology and differentiation as well as the tumor T cell composition will aid the further identification of specific T cell subsets for targeted cancer therapy, further reducing adverse effects.
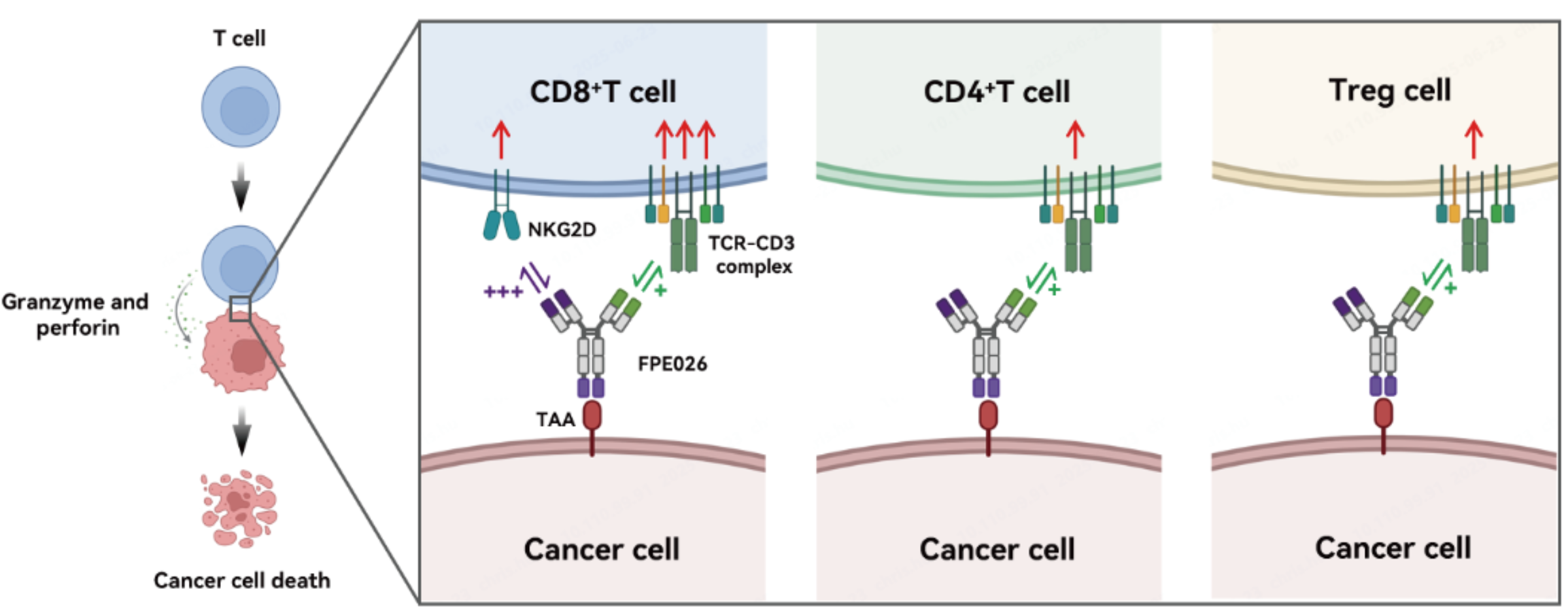
FPE026: a tri-specific TCE CDH17×CD3×NKG2D
Guangdong Fapon Biopharma Inc. is developing FPE026, a next-generation CD8-selective tri-specific T cell engager targeting CDH17. FPE026 is a CD8-biased TCE strongly activates CD8+T cells while minimally activating CD4+T cells and Treg cells, thereby reducing toxicity and improving therapeutic efficacy. Selective CD8+T cell activation is based on NKG2D that is highly expressed on CD8+T cells but is rarely expressed on CD4+T cells and Tregs, providing inherent selectivity for CD8+T-cell activation.
TCEs targeting the TCRαβ domains on T cells instead of CD3 have emerged as a novel approach
Adimab: Pan-αβ-T-cell reactive anti-TCR VHH bsAbs
Adimab is developing bispecific TCEs incorporating αTCR VHHs as an orthogonal and broadly applicable platform for T cell redirection. Their αTCR VHH lineages are targeting the TCR constant domain and occupy a distinct epitope bin, engaging both TRBC1 and TRBC2 for pan-⍺β T cell redirection. Molecular format or geometry of their αTCR VHH bispecific TCEs influences activity, with N-terminal bispecific formats showing higher potency than C-terminal formats.
Their data demonstrate that unlike CD3-based TCEs, where CD3 affinity strongly influences potency, αTCR-based TCEs retain efficacy even with lower-affinity TCR arms, with potency more dependent on the tumor antigen (TAA) arm. The ability to maintain potency while decoupling affinity from the TCR-targeting arm expands the design space for TCEs and supports generation of bi- and multispecifics with a broad therapeutic window.
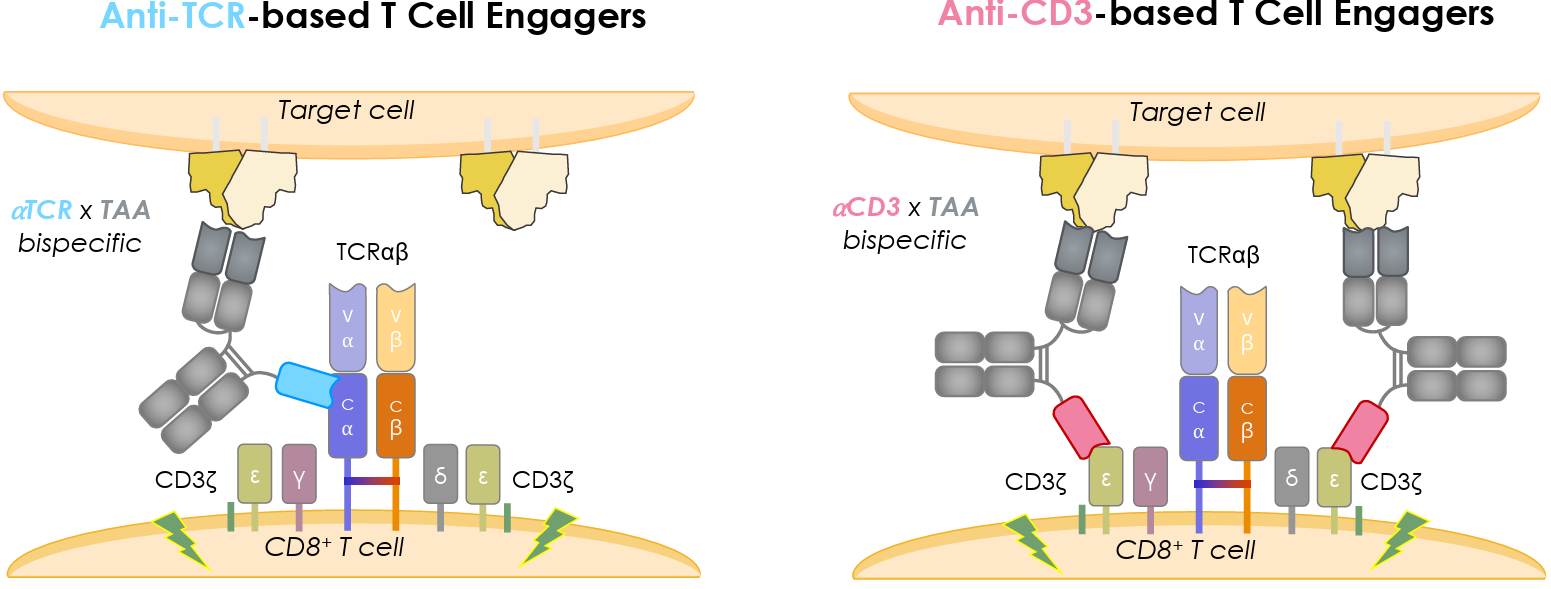
Pan-αβ-T-cell engagers by Adimab are targeting the TCR constant domain as an alternative engagement node (ref. Adimab.com Resources)
The CD33/CD123 nanobody TCE developed by Sanofi validates the anti-TCRαβ VHH platform. CD33/CD123 - TCE effectively eliminates acute myeloid leukemia cells in mice and targets cells in nonhuman primates without causing systemic cytokine upregulation linked to CRS. Their results (published in https://doi.org/10.1182/bloodadvances.2023011858 ) highlight the promise for clinical applications of TCEs using an alternative T cell engaging strategy.
AZD5492 and AZD8359: CD8-guided TCEs for B-Non Hodgkin lymphoma and Prostate cancer, respectively
Astra Zeneca is currently evaluating a first-in-class CD8-guided TCE AZD5492 in FIH study in patients with R/R B-Cell Malignancies. AZD5492 is a trispecific antibody which harbors two Fab binding domains to CD20, one VHH binding domain to TCR, one VHH binding domain to CD8 co-receptor.
At AACR2026, Suzanne Sitnikova PhD, of AstraZeneca, Cambridge in the United Kingdom, introduced AZD8359, a novel CD8 biased TCE targeting the novel prostate cancer antigen STEAP2. AZD8359 treatment induced antitumor activity in pre-clinical models and caused reduced cytokine release compared with conventional T-cell engagers.
By preferentially engaging CD8+ T-cells rather than CD4+ T-cells, AZD5492 and AZD8359 aim to eliminate cancer cells while minimizing immune-related toxicities (i.e. cytokine release by CD4+ cells) commonly associated with current T-cell engagers. With the optimal CD8-guided immune synapse formation Astra Zeneca is aiming to achieve widest clinical therapeutic index and they are currenlty evaluating 2 assets in clinical trials NCT07192614 and NCT07529717.
Trispecific Selective T cell Activation Repertoire (tri-STAR) platform by Marengo Therapeutics
Another example is the trispecific Selective T cell Activation Repertoire (tri-STAR) platform (MarengoTherapeutics), which contains a tumor-targeting domain and selectively activates T cell subsets that are enriched in solid tumors via specificity to TCR variable beta (Vβ6/Vβ10)-chains. This approach allows for tumor-directed cytotoxicity but only leads to the activation of a small proportion of T cells, thereby reducing the potential for CRS.
The tri-STAR platform builds upon the initial approach of selectively expanding a subset of T cells with an anti-TCR Vβ6 and Vβ10 antibody fused to interleukin-2 (IL-2) molecule. (refs: 10.1126/sciadv.adj6174 and 10.1126/scitranslmed.adi025 ).
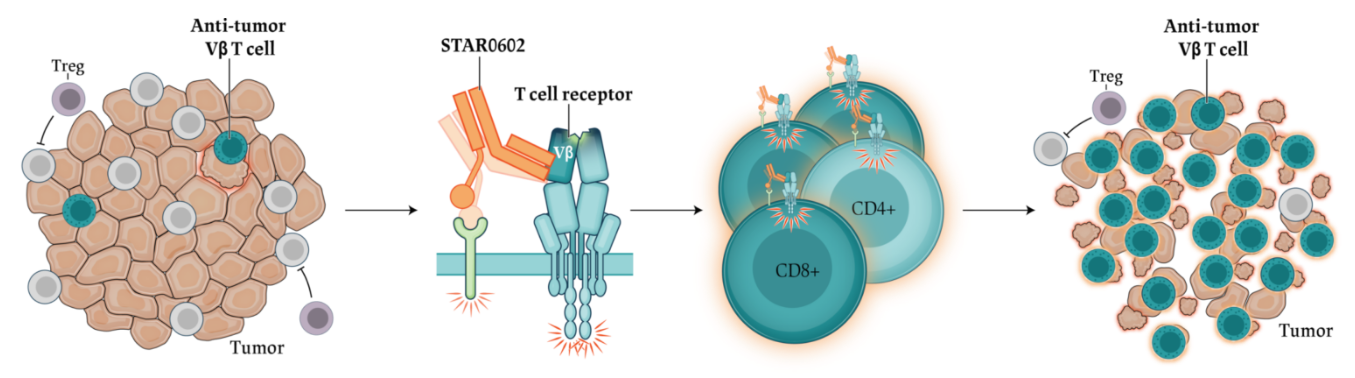
Invikafusp alfa (STAR0602) is the first dual T cell agonist generated from Marengo’s STAR™ platform delivering two activation signals to a targeted subset of the T cell populations. It is designed to selectively activate Vβ6/10 T cells, a common subset of T cells highly prevalent in tumor infiltrating T cells (TILs) found in all cancers by combining a non-clonal mode of TCR activation with a T cell co-stimulatory signal in a single molecule. This innovative approach promotes the expansion of clonally diverse, effector memory Vβ 6/10 T cells, enhancing anti-tumor immunity and enabling durable tumor clearance.
The activation of viral and/or memory CD8+ T cells, while minimizing systemic T cell activation and CRS
AGB101: Vβ17 x DLL3 by Agni Bio
Agni Bio is taking TCEs a step further with increased precision in engaging a specific T memory subpopulation whose unique features enable a higher therapeutic index and more durable responses. Agni is advancing Vβ17 TCE platform with their front runner AGB101 as a best in class DLL3 targeting agent. By redirecting a cytotoxic T memory subpopulation less prone to exhaustion, AGB101 shows reduced cytokine release in preclinical studies, and is expected to have a greater therapeutic index.
Following natural influenza A infection, TCR Vβ17+ CTL dominate the response against infection (to the extent that only one of nine adult CTL lines generated retains any functional activity after in vitro depletion of Vβ17+ CTLs). These TCR .Vβ17+ CTLs persist in healthy individuals and cancer patients and present a consistent subset of memory population primed to be re-engaged. https://doi.org/10.1093/intimm/13.11.1373
AGB101 is part of Agni Bio’s Vβ17 platform of TCR-directed bispecific antibodies designed to improve the precision and effectiveness of immune engagement in cancer and autoimmune therapies. AGB103 (a Vβ17 x CD19) is currently in lead optimization phase.
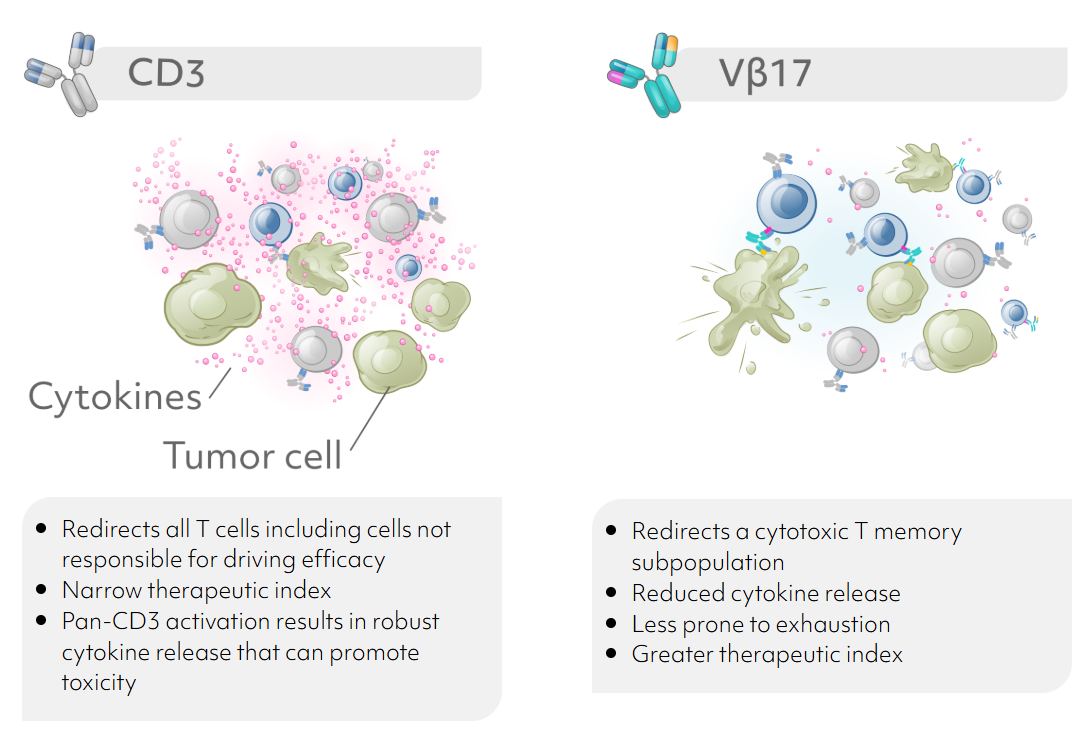
Key highlights of Vβ17 platform developed by Agni Bio (ref. Agni Bio Science)
Concluding remarks
Where is the future taking us: ideally we will identify additional TCRs associated with effector CTL phenotype (i.e. CX3CR1+ Teff) and generate bsAbs targeting those Vβ TCRs.
From there, the additional improvement in activity can be achieved with combining TCEs with complimentary therapies. Advancements in TCE targeting have significant implications for the potential of combination therapies. TCEs with enhanced therapeutic windows allow for more aggressive and effective combination strategies, such as pairing with checkpoint inhibitors, chemotherapy, tyrosine kinase inhibitors, or antibody-drug conjugates. Safe combination therapies will be especially beneficial in solid tumors where the TME often presents a major challenge.



