Bridging T cells to Tumors: CARs vs BsAb
The latest generation immunotherapeutics are designed to harness the potent tumor-killing capacity of T cells. CAR T-cell therapies and bispecific antibodies (BsAb) that redirect T cells to CD19 have generated fantastic responses in B-cell malignancies and revolutionized the field of cancer immunotherapy in the past few years (see section 6 in the blog recaping 2017) by demonstrating that redirection of T cells against tumors holds much promise for the treatment of cancer. The advent of these novel T cell-engaging platforms has been enabled by the single-chain variable fragment (scFv) technology. Several versions of T cell therapies hold a great potential: CAR and TCR products as well as bispecific recombinant proteins (BsAb) redirecting the activity of T cells against tumor targets (BiTE, ImmTAC, DART, Diabody).

Figure 1. Advantages and disadvantages of some adoptive cell therapy approaches.
Recent advances and positive clinical results from BiTE and CAR T-cell therapies have generated hope and excitement, however both therapies are facing similar dilemmas,
such as toxicity concerns in hematologic cancers and a lack of response in solid cancers.
CARs
A Chimeric antigen receptor (CAR, also known as chimeric immunoreceptor, chimeric T cell receptor, artificial T cell receptor or CAR-T) is a synthetic receptor comprising an extracellular tumor antigen recognition domain, which is fused to the CD3ζ chain for intracellular signaling and contains one or more costimulatory domains. The binding of scFv to tumor cells triggers the T-cell receptor (TCR) intracellular domain and leads to T-cell responses against antigen expressing cells.
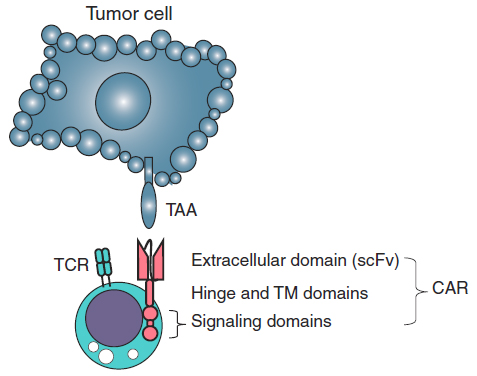
Figure 2. The design of a CAR T cell. A CAR comprises an extracellular
domain, which is an scFv targeting TAA. The scFv is followed by a hinge of
varying length and flexibility (CD8 as an example) and various combinations of endodomains that provide T-cell activation signals, such as CD3ζ, and a costimulation signal, such as CD28. TM, transmembrane. (Slaney et al., 2018)
In 2017, the Food and Drug Administration (FDA) approved the use of two CAR T-cell therapies. Both use genetically modified cells that recognize a protein (called CD-19) on the surface of cancerous B cells.
- Novartis was the first to launch a CAR-T therapy, called Kymriah (tisagenlecleucel ), last August (priced at $475,000). Kymriah is for pediatric and young adult patients age 25 or younger with B-cell acute lymphoblastic leukemia (ALL). It is a one-time treatment that has shown an 83% remission rate after three months in clinical trials with patients that do not respond to standard treatments. However, 49% of the patients suffered strong cytokine release syndrome (CRS). This side effect has been responsible for several deaths in clinical trials run by Novartis’ competitors. For example, Juno Therapeutics had to terminate its lead CAR-T program last March after a total of 5 patients died of cerebral edema caused by the therapy.
- Gilead was the second to bring a CAR-T therapy to the market. The big pharma acquired Kite Pharma just weeks before the FDA approved its CAR-T cell therapy, Yescarta (axicabtagene ciloleucel, priced at $373,000). The therapy induced remission in 72% of patients with aggressive B-cell non-Hodgkin lymphoma that has relapsed or does not respond to standard treatments. However, the trial reported three deaths linked to the side effects of the therapy.

Figure 3. The history of CAR therapy development.
Other companies running clinical trials with CAR-T therapy include Juno Therapeutics, Celgene and Mustang Bio in the US, Celyad in Belgium and the French Servier in partnership with Pfizer. Cellectis and Celyad are developing their own allogeneic CAR-T cells each, which has created friction between them. Cellectis has been the first to test two of these off-the-shelf CAR-T cells in clinical trials — one of them now licensed to Servier and Pfizer. For its part, Celyad is still in the preclinical stage with its allogeneic CAR-T cells, which have been licensed to Novartis.
Various molecular formats of CAR have been developed, differing in their extracellular, transmembrane, and cytoplasmic domains. The first CAR T cells are comprised of a high-affinity scFv fused to the hinge and transmembrane domains of either CD8 or CD28, and the intracellular domains of the co-stimulatory molecules 4-1BB or CD28, followed by CD3ζ. Inclusion of at least one costimulation domain is critical to allow the CAR T cells to expand and persist and effectively kill tumor cells in vivo. Some costimulatory molecules, such as 4-1BB, may be better than others in certain respects, such as facilitating long-term persistance.

Figure 4. Chimeric antigen receptor design. First generation CARs include the intracellular domain of CD3ζ, which contains 3 ITAM domains (red). Second generation CARs also include the intracellular domain of a costimulatory molecule such as 4-1BB or CD28, whereas third generation CARs include 2 or more costimulatory domains in addition to CD3ζ. (Zhukovsky et al. 2018).
For solid tumor targets low-affinity scFv are being employed to achieve specificity towards tumor cells expressing high levels of target.

Figure 5. Solid tumor targets amenable for CAR-T cell approaches.
Some investigators have designed CAR T cells specifically targeting the tumor microenvironment to overcome the immunosuppressive effect. For example, chimeric cytokine receptors composed of the extracellular domain of the IL4R fused to the intracellular domain of the IL7R (to provide signal 3 for T cell support) leads to activation and proliferation of T cells in the presence of the normally inhibitory cytokine IL4. Further, CAR T cells have been modified to express CCR2b, the chemokine receptor for CCL2 that is highly expressed by the target tumors. To facilitate the penetration into tumors, construction of CARs targeting VEGFR2 or αvβ3 integrin overexpressed on tumor vasculature has been reported. Alternative approach to modulate the functional barrier preventing infiltration of endogenous effector T cells is to use CAR T cells as carriers/supply of certain cytokines or co-stimulatory molecules, such as IL-12 and CD40L.
BsAbs
The other area that has a high level of excitement for using scFv technologies is the BsAb approach. In the mid-1980s, the first BsAb that specifically redirected T cells to a target antigen was generated. Since then, the BsAb field has gradually expanded, with >2,500 publications in PubMed in 2017. Although proof-of-concept studies were performed by chemically crosslinking 2 monoclonal antibodies (mAbs) to generate BsAbs, the majority of BsAbs are currently generated by recombinant DNA technology. Numerous BsAb formats to redirect T cells to tumor antigens have been generated and are undergoing preclinical and clinical testing (detailed review of >100 BsAb formats by Brinkman and Konterman, 2016).
By bridging T cells and target cells with a BsAbs, T-cell activation is major histocompatibility complex (MHC) unrestricted and no longer depends on the native T-cell receptor specificity of the activated T cell.
Although it has been shown for a particular antigen that one BsAb format is superior to others, one size will most likely not fit all, because there is an intricate interplay among MAb affinity, epitope location within the targeted antigen, and antigen density and mobility on the target cell surface, all of which contribute to optimal T-cell activation.
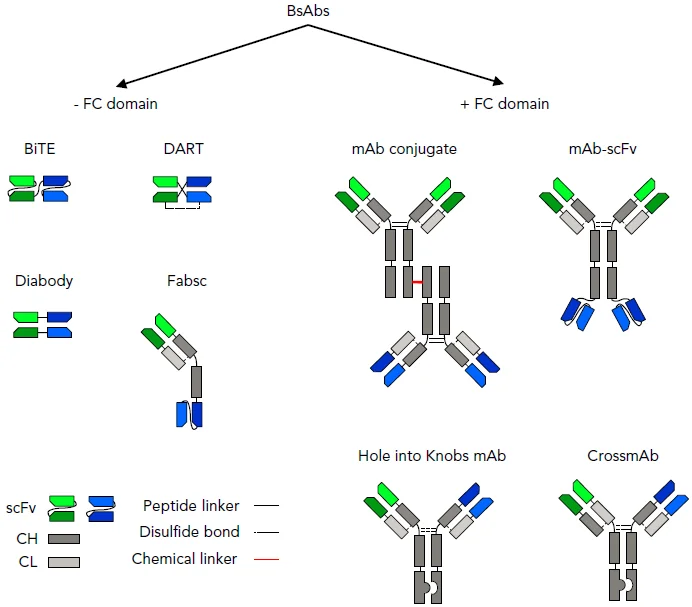
Figure 6. Selected BsAb formats. BsAbs can be broadly divided into molecules that contain or do not
contain an immunoglobulin G backbone with a functional Fc domain. BsABs can be created by chemical crosslinking 2 mAbs or recombinant DNA technology. Fab, fragment antigen binding; CH, heavy chain; CL, light chain; DART, dual-affinity retargeting; sc, single chain; scFv, single-chain variable fragment (Velasquez et al., 2018).
Among BsAbs, bispecific T-cell engagers (BiTEs) have garnered particular interest. BiTE is a recombinant bispecific protein that has two linked scFvs from two different antibodies, one targeting a cell-surface molecule on T cells (for example, CD3ε) and the other targeting antigens on the surface of malignant cells. The two scFvs are linked together by a short flexible linker. The CD19-specific BiTE Blincyto® (blinatumomab) has shown impressive clinical results for CD19-positive B-cell malignancies, resulting in its US FDA approval for the treatment of Philadelphia chromosome-negative relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL) in 2014. Blinatumomab comprises an anti-CD19 scFv in the VL-VH orientation linked through a G4S linker to an anti-CD3 scFv in the VH-VL orientation (DrugBank entry DB09052). Because of the small size, BiTE molecules are rapidly cleared from circulation with a terminal half-life of ∼1.25h. The antibody is thus administered as continuous intravenous infusion at a constant flow rate with a portable pump in repeated four-week cycles.
Tandem scFv that include an anti-CD16 scFv, i.e., bispecific killer cell engagers (BiKEs) were further developed for retargeting of natural killer (NK) cells.
Diabodies (Db) are bivalent molecules composed of two chains, each comprising a VH and VL domain, either from the same or from different antibodies. In the diabody format, the two variable domains are connected by a short linker that is usually 5 residues, e.g., GGGGS. Because the linker length is substantially shorter than that required to allow intrachain assembly of an antigen-binding site, which would result in a scFv, two chains dimerize in a head-to-tail orientation resulting in a compact molecule. Expressing two chains within the same cell, with either configuration VHA-VLB and VHB-VLA (A and B representing two different specificities) or VLA-VHB and VLB-VHA, results in bispecific heterodimers with correct pairing of the corresponding variable domains. The issues with bispecific Db are the possibile assembly of nonfunctional homodimers with incorrect pairing of the variable domains (VHA/VLB, VHB/VLA) and instability due to noncovalent assembly of the two chains. An alternative applied to diabodies is the covalent linkage of the two chains through C-terminal cysteine residues. This was used to generate so-called dual-affinity retargeting (DART) proteins.
One of the major hurdles for BiTEs is their short half-life in serum. After injection, BiTEs are rapidly catabolized and cleared from the circulation, leading to a short serum half-life of around 2 hours. To overcome the limitation, current approaches include attaching BiTEs to heavy-chain fragments or other novel designs to increase the serum life. The other major hurdle for treating solid tumors using BiTE is the density and types of T cells already in the tumor bed, as well as whether preexisting T cells in the tumor may already have an exhausted phenotype or may be Tregs.
Recent enthusiasm surrounding BiTEs extends toward their production in vivo. One strategy is to produce genetically modified T cells (ENG-T cells) for making BiTE to overcome the limitations of BiTEs’ short half-life and passive biodistribution. These ENG-T cells expanded in vivo and increased the production of BiTEs after activation, obviating the need for continuous infusion of engager molecules. Another recent promising strategy is to use an oncolytic virus engineered to express a BiTE.
Key attributes of mechanism of action

Figure 7. Comparison of CAR T cells and BiTEs
T cell subsets: In the CAR T-cell approach, CD4+ T cells and CD8+ T cells are usually mixed and genetically modified with CAR and infused back to the patients in clinical trials. Although there is no doubt that CD8+ CAR T cells are the major players in killing cancer cells, CD4+ T cells armed with a CAR can also be activated via CAR and showed cytolytic activities. Similarly, it has been demonstrated that when incubated with BiTEs, both CD8+ and CD4+ T cells can be activated and induce target tumor cell death, with CD8+ cells killing faster than CD4+ T cells.
T cell phenotype: It is now evident that less differentiated CAR T cells have better efficacy in vivo. Therefore, although TEM and TEFF can be present at high frequency at the peak of the anticancer immune response, their persistence is poor. Accordingly, strategies favoring the generation and preservation of TSCM and TCM have been applied in the CAR T-cell therapy field. In contrast, in BiTE treatment antigen experienced T-cell subsets mediate BiTE-induced tumor cell death, whereas naïve T cells are not activated when engaged by BiTE. Therefore, T cell populations independent of co-stimulatory signaling are the major contributor to the cytotoxic potency of BiTE.
Linker domain: Differences exist between the synapse formed between T cells and target cells when mediated by BiTEs compared with CARs, which potentially arises because BiTEs engage the natural CD3 complex, whereas CARs do not. Both, however, can induce serial killing of cancer cells. The distance between the TAA epitope and the target cell membrane determines the activity of the BiTE and may explain the differences in the efficacy between different BsAb formats.
Expansion of T cells: CAR T cells have been demonstrated to expand up to, or even exceeding, 1,000-fold and to reach greater than 20% of circulating lymphocytes. In the case of BiTEs, a large pool of antigen-experienced T cells can be exploited to provide substantial numbers of redirected T cells, with less reliance on expansion. Increases of the order of 2- to 4-fold in circulating T cells have been described. The provision of large numbers of tumor-reactive T cells in the solid-tumor setting presents different challenges for the BiTE and CAR T-cell approaches. Whereas extensive expansion and prolonged persistence has been demonstrated for CAR T cells in hematologic cancers, little if any expansion of
CAR T cells is usually demonstrated when treating solid cancers. Because the BiTE approach is potentially less reliant on T-cell expansion, it may be more successful against solid tumors. However, the success of this approach is dependent on the sustained loading of BiTEs on T cells, which may be compromised during the process of penetration of solid tumors.
Valency and affinity: Initially, low CD3 affinity (KD ~ 10-7 M and higher) was considered a requirement for avoidance of non-tumor-target- specific activation of T cells. It was suggested that high affinity (KD ~ 10-9–10-8 M and lower), bivalent
binding to CD3 might lead to non-target-specific T cell activation. However, subsequent reports suggest that non-target-specific triggering of T cells depend on cross-linking or immobilization of the antibody by FcgR+ cells, and are nota direc function of bivalency.
Toxicity
CAR T-cell and BsAb therapies can be associated with some toxicities.
The most commonly observed toxicity in these therapies is cytokine release storm (CRS), a systemic response due to elevated levels of cytokines, such as IL6 and IFNγ. The patients with highly elevated levels of IL6 are the ones with maximum CAR T-cell activation and proliferation and anti-IL6R Ab (tocilizumab) can reverse the life-threatening CRS without inhibiting CAR T-cell treatment efficacy. Neurotoxicity, also termed CAR T cell–related encephalopathy syndrome (CRES), has also been reported and its pathogenesis remains unclear. CRES is generally reversible using anti-IL6R Ab and/or corticosteroids. Neurologic adverse effects are also among the most frequent adverse effects in BiTE therapy and patients are required to be premedicated with dexamethasone. B-cell aplasia is associated with CD19 CAR T-cell and blinatumumab therapies, because CD19 is expressed on normal B cells, but injections of immunoglobulin can be used to maintain levels of circulating antibodies.
Future of T cell redirection therapies
The main improvement strategies are focused on:
- enhanced anti-tumor clinical activity by either providing positive stimulation or immune checkpoint blockade (co-therapies)
- novel targets (to overcome tumor microenvironment-imposed resistance in the solid tumors and acquired resistance in haematological malignancies due to downregulation/loss of the antigen expression)
- novel sources of T cells (allogeneic off-the-shelf T cell therapies)
- fine tuning of the effector cells (to limit the on-target toxicity)
One must wonder how it became initially accepted that the new CD3-engaging BsAb obviated the requirement of the second signal to mobilize all possible T cell populations. It is now understood that this is not the case, and that memory T cells are the major
contributors to the cytotoxic potency of BiTEs and perhaps the other T cell redirecting BsAb; in the future realizing the full clinical potential of these molecules may require inclusion of costimulatory modalities.
Advanced CAR engineering techniques aim to expand the possibility of targeting multiple antigens, by using CAR-T cells that will recognize two antigens and only become activated by the correct combination. In such case the first antigen delivers signal 1 with a first generation CAR and the second antigen delivers signal 2 by engaging a chimeric co-stimulatory CAR encoding only costimulation without CD3ζ. Similary, a dual TAA-targeting strategy for the specific optimization of T cell redirecting applications for solid tumors may enhance the selectivity and safety of T cell redirecting
antibodies and broaden their range of clinical indications. In this approach, the affinity of a trispecific T cell redirecting antibody for each TAA is selected low to minimize binding to healthy tissues expressing a single antigen, whereas the binding to cancer cells, which express both TAAs, is substantially increased due to avidity. For this
approach to succeed, the selection of TAAs should be limited to those whose expression is known, or expected, to be in close cell-surface proximity. Finally, this approach
may also improve clinical efficacy of T cell redirecting antibodies since targeting two TAA may also reduce tumor escape mechanisms.
Novel recent CAR design has a backbone of a CAR, but with the extracellular domain of CD16 rather than a scFv and the antigen specificity is conferred by binding of a soluble anitbody to CD16. Similarly, Celyad developed a version of CAR-T (Celyad’s CAR-T therapy called CYAD-01) that makes engineered T cells expressing natural killer receptor (NKG2D) that targets eight different tumor ligands simultaneously instead of a tumor-specific/associated antigen.
Bluebird bio, in partnership with Scottish biotech TC Biopharm, is also targeting solid tumors with an improved version of CAR-T that uses a specific class of T cells known as gamma delta T cells. They selectively target cancer cells without attacking healthy cells, which could significantly reduce the toxicity of the therapy.
Another novel design is based on a drug-activated CAR, where the signaling components of the CAR are divided among different proteins that only come together in the presence of a drug that causes them to dimerize and signal. Cellectis, in France licensed its CAR-T Therapy UCAR19 to Servier and Pfizer. UCART19 includes a switch control system that only activates the engineered T cells when the patient is given the drug rapamycin. The therapy is in Phase I, after saving two babies with aggressive forms of leukemia in compassionate cases. Bellicum Pharmaceuticals, in the US, is developing a similar technology called GoCAR-T that requires the drug rimiducid for CAR-T cell activation.
Apart from regulating the activation, there is great interest to develop ways to terminate CAR T cell function to limit the short-term or long-term toxicity. 'Suicide genes' which can be introduced in a bi-cistronic vector along with the CAR, allow selective destruction of expressing cells in the face of unacceptable toxicity by administration of an activating pharmaceutical agent. Several suicide genes have been described. One family of suicide genes, such as RQR8 and huEGFRt, are surface proteins recognized and depleted by cognate therapeutic mAbs. Other suicide genes have been described that are activated by a small molecule. Two of these have been tested in clinical studies (of haploidentical HSC transplantation): herpes simplex virus thymidine kinase (HSV-TK) and inducible caspase 9 (iCasp9). Expression of HSV-TK in T cells confers susceptibility to ganciclovir. HSV-TK is a highly effective suicide gene strategy, but immunogenicity limits application to clinical settings of profound immunosuppression. iCasp9 is a fusion of a mutated FKBP12 with the catalytic domain of caspase 9. The FKBP12 is mutated to allow docking of a small molecular chemical inducer of dimerization (CID, AP1903/AP20187) that cannot bind wild-type (WT) FKBP12 and is, hence, otherwise pharmacologically inert. Because iCasp9 is a fusion of self-proteins, it is unlikely to be immunogenic.
Concluding remarks: CARs vs BsAb direct comparison
A few studies directly compared the functional efficacy of the two strategies in their antitumor effect. An early study compared the CAR and BiTE treatment antitumor response in vitro using the same anticancer scFv against a murine fibrosarcoma epitope, 237. Although both strategies enhanced T cell–mediated antitumor effect, CAR T cells were more potent than the BiTE approach against cancers expressing lower levels of antigens (Stone et al., 2012). In a recent study, BsAb (specific for both CD3 and GD2), was compared with a CAR that contained the same anti-GD2 scFv (Hoseini et al., 2017). For treating the GD2+ tumors in immunocompromised mice, BC119 with untransduced T cells conferred rapid shrinkage of the established tumors, whereas CAR T cell–treated tumors showed a delayed regression for 2 weeks. The percentage of
tumor-infiltrating lymphocytes (TIL) in the BC119 treatment was higher than that in the CAR T-cell treatment. The number and composition of TILs could partially explain the superior antitumor effect mediated by BiTE over CAR T-cell treatment. It is noteworthy that the above study used immunocompromised mice with adoptive transferred T cells, whereas in the clinic, BiTEs are usually administered without adoptive transfer.
In the clinic, it is possible that patients refractory to one therapy respond to the other. For example, 2 patients refractory to blinatumomab achieved CR after CD19-CAR treatment (a trial run by Maude and colleagues in 2014), suggesting that a lack of response to one immunotherapy against the same target may not preclude a successful therapy outcome with another, even with the same target.
In addition, there could conceivably be some benefit from using CAR T cells and BiTEs simultaneously against tumors. A preclinical study targeting the alpha folate receptor using a CAR, while also targeting EGFR using an oncolytic virus encoding a BiTE, led to increased inhibition of tumors (Wing et al., 2018).