
Epigenetics meets immuno-oncology
Cancer immunotherapies, including checkpoint inhibitor therapy and adoptive cell therapy (ACT), can induce striking clinical responses in cancer patients, although responses are limited to a subset of individuals within specific cancer subtypes. Many human cancers are resistant to immunotherapy targeting T-cell mediated cytotoxicity. While multiple factors are responsible for incomplete effectiveness, two major roadblocks include the development of T cell exhaustion and the functional impairment of cells used for ACT.
As highlighted in a recent review by Henning et al. (2018), epigenetic processes direct a substantial portion of the T cell differentiation programme together with lineage-specific transcription factors. Alterations to gene expression affecting T cell differentiation and function - achieved by targeting epigenetic enzymes or the functionally relevant genomic loci that they regulate - would eliminate the negative side effects of more indiscriminate genetic or pharmacological interventions and have the potential to substantially improve current immunotherapies by promoting memory cell differentiation or by preventing or reversing T cell exhaustion.
1. Fundamentals of epigenetics in cancer
The term ‘epigenetics’ refers to the control of gene transcription by mechanisms that are heritable through cell division but not related to the genetic code, DNA. Epigenetic marks tell RNA how to use the code for protein synthesis. The protein machinery that adds, removes or recognizes these marks are called writers, erasers, and readers (Figure 1).

Figure 1. Chromatin regulators involved in transcription induction. List of chromatin remodellers, chromatin modifiers (writers and erasers of histone posttranslational modifications (PTMs)) and histone PTM readers. Their function and status in cancer are indicated (Marazzi et al., 2017).
The 3 major epigenetic mechanism are depicted in Figure 2.

Figure 2: Major epigenetic mechanisms affecting gene activity (Rajender et al., 2011).
Some marks are inherited but some are acquired; the cells can send signals to modify these epigenetic marks that will allow the correct proteins to be synthesized for the correct environment. So, though cells share identical genetic information, the phenotype and function are determined by adaptable and heritable gene expression patterns in response to the environment. Interestingly, RNA and proteins can also be directly modified by epigenetic modifiers.
In a concise sketch video below Drs. Clark and Patterson (Garvan Institute of Medical Research) highlight key messages about the role epigenetics plays in cancer. For a more comprehensive overview of the cancer epigenetics field listen to Susan Clark's lecture from 2013.
2. The role of epigenetics in immune cell function
The immune system consists of a highly responsive and stably adaptive (bearing the memory capacity) network of interacting cells. Epigenetic regulation of gene expression plays a key role in this functional flexibility. Therefore, epigenetic targeting may be beneficial in cancer immunotherapy by reversing immune avoidance and escape mechanisms employed by cancer cells, as well as by modulating immune cell differentiation and function.
Recently, all 3 major epigenetic mechanisms have attracted attention for their role in directing the immune cell function. 4 types of inhibitors of these processes are currently explored (pre)clinically in this context:
· DNMTi: inhibitors of DNA methyltransferases (writer).
· HDACi: inhibitors of histone deacetylases (eraser).
· EZH2i: inhibitors of EZH2, a histone methyltransferase for H3K27 (writer).
· Chromatin remodelling inhibitors (e.g. BETi, bromodomain inhibitors of BET proteins, readers of histone acetylation).

Figure 3. Chromatin remodelling and susceptibility to inhibitors. Chemical inhibitors of regulatory proteins that control gene activation preferentially affect the expression of genes that require chromatin remodelling for induction in response to inflammatory or mitogenic stimuli. (a) Illustration of drug inhibitors acting on their respective targets to affect inducible gene expression. Inhibition (denoted by an ‘i’) of chromatin factors involved in RNA polymerase II (Pol II) pause–release, such as bromodomain and extra-terminal domain (BET) proteins and cyclin-dependent kinases (CDK7, CDK8 and CDK9), primarily affect genes with promoters that are linked to super enhancers and more in general to highly-induced genes. Inhibition of protein arginine N-methyltransferases (PRMTs) at R-loops and inhibition of DNA topoisomerase 1 (TOP1) also preferentially affect inducible genes such as those linked to the inflammatory response. (b) Chromatin features of chromatin-remodelling-independent genes and chromatin-remodelling-dependent genes. (c) Chromatin regulators and examples of their inhibitors (Mazzini et al., 2017).
Inhibition of epigenetic regulation of gene transcription by covalent modification of DNA or histones has been shown to lead to following processes:
· Re-expression of MHC molecules to increase the availability of tumor antigens
· Increased surface expression of calreticulin thereby facilitating antigen uptake
· Activation of interferon signaling genes
· Alteration of cytokine production
· Demethylation of endogenous viral sequences that triggers dsRNA sensors and an IFN response
· Blockage of CD4+ T cell conversion into Treg cells
· Reversion of exhausted T-cells
· Reduction of MDSC build up
· Enhancement of cell surface expression of immune checkpoints and co-stimulatory molecules
IFN-related genes, in addition to others key genes that are re-activated in response to different epigenetic treatments (including TAAs, APM components, MHC class I/II molecules, co-stimulatory/accessory molecules, chemokines, and immune checkpoints), can be used to determine the impact of epigenetic therapies on re-education of tumour cells to become more visible for immune attack.
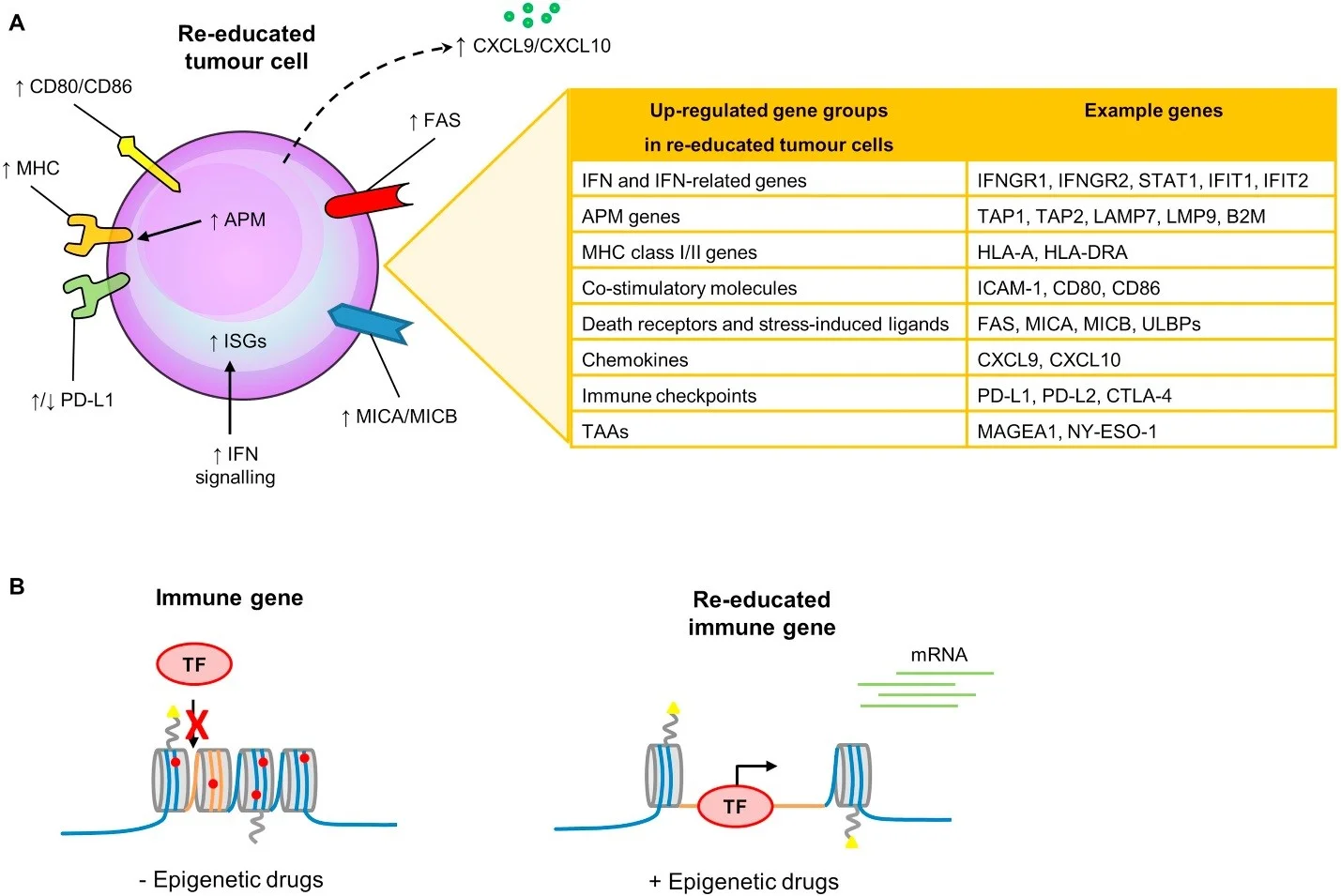
Figure 4. Re-education of cancers cell towards visibility for immune attack (Dunn and Rao, 2018). (A) Several key papers have recently identified IFN signalling genes (1) that are activated in response to DNA demethylation (Roulois et al., 2015 and Chiappinelli et al., 2016) or (2) that have genomic defects in immune checkpoint resistant tumours (Gao et al., 2016). (B) At the chromatin level, immune genes are silenced in a closed heterochromatin state in tumour cells and the addition of epigenetic drugs re-educates immune genes to become open and transcriptionally active (Dunn and Rao, 2018)
3. Combining epigenetic modulation with immune checkpoint blockage
With the recent success of immunotherapy and the introduction into the clinic of novel targeted epigenetic drugs, it is paramount to carefully evaluate the therapeutic potential of such combinations. By acting on the tumour as well as on the tumour microenvironment, epigenetic modifiers have the potential to sensitize tumours to subsequent adoptive T cell therapy or to combinations of immune checkpoint inhibitors.
While anti-PD1 allows for T-cell activation in situ by blocking the inhibitory interactions between PD1 on T-cells and its ligands PD-L1 and PD-L2 on tumor cells and antigen presenting cells in the tumor microenvironment, epigenetic modifiers can suppress immune checkpoint (IC) expression on tumor cells and in the tumor microenvironment (BETi). For example, JQ1 can inhibit the expression of leukocyte
surface antigen CD47 and PDL1 in models of ovarian cancer and in MYC-driven tumours.

Figure 5. The differences in action of epigenetic modulation on anti-CTLA4 and anti-PD1 IC therapy (Gallagher et al., 2017). (A) Anti-CTLA4 (αCTLA4) enhances T-cell priming in draining lymph nodes by blocking the inhibitory B7-CTLA4 interactions while permitting the co-stimulatory B7-CD28 engagement. B7 expression on dendritic cells is increased as it is controlled by Treg-cells in a CTLA4 dependent fashion. DNMTi increase the availability of tumor antigens, including neo-antigens, while HDACi improve antigen uptake, thus facilitating priming of novel tumor-specific T-cell clones. (B) Anti-PD1 (αPD1) activates T-cells at the tumor site and its efficacy depends upon the presence of tumor immune infiltrate including CD8 T-cells but also CD4 T-cells and antigen-presenting cells such as tumor-associated macrophages and dendritic cells. Antigen persistence and chronic inflammation render tumor-infiltrating T-cells dysfunctional through epigenetic modifications and also convert CD4 T-cells into immunosuppressive Treg-cells.
Treatment of tumour cells with DNMTi elicits a favourable and potent interferon response in cancer by inducing the expression of double-stranded RNA endogenous retroviral elements (ERVs). Transcripts of ERVs and satellite repeats and aberrant transcription from repetitive elements that mimic viral RNA stimulate innate pattern-recognition receptors and induce inflammation (Roulois et al., 2015; Chiappinelli et al., 2015). Interestingly, a similar DNMT1‑mediated effect has also been observed upon inhibition of the cell cycle kinases CDK4 and CDK6.
Additional benefits of DNMTi include the reactivation of major histocompatibility complex class I genes, which are often transcriptionally silenced in cancer. Besides, DNMTi increase tumor antigenicity by re-expression of tumor antigens such as the cancer-testis antigens, and altering cytokine production.
The potential of targeting tumour microenvironment cells is further demonstrated by the treatment of lung adenocarcinomas with a selective inhibitor of HDAC6 (ricolinostat), which supports the activation of T cells and improves the function of antigen-presenting cells. The combination of ricolinostat with JQ1, which instead attenuates the immune suppressive functions of CD4+FOXP3+ T cells, synergizes to facilitate immune-mediated tumour growth arrest, leading to improved survival and tumour eradication. Similarly, EZH2 inhibitors block CD4 T-cell conversion into Treg-cells.
Other encouraging preclinical studies have combined DNMTi with HDACi to reduce circulating myeloid-derived suppressor cells and to enhance the effect of immune checkpoint blockade.
More recently, the epigenetic reprogramming of exhausted CD8+ T cells has been found to be a limiting factor in long-lasting effective treatment with PD1 blockers (Sen et al., 2016; Pauken et al., 2016). Anti‑PD1 treatment cannot fully reprogramme exhausted cells to reinstate the functions of memory T cells or effector T cells, but it could rewire and reactivate the transcriptional network of key inducible immune target genes (NF-kB, IRF1, IRF2, ..). The therapeutic implication of these studies is that treatment with epigenetic drugs could be used to selectively expand long term memory-like cells, improve the reinvigorating effects on CD8+ T cells induced by immune checkpoint blockade or curb the unwanted side effects of hyper-activation of the immune system. Henning et al., 2017 have recently examined the translational potential of epigenetic interventions to improve CD8+ T cell function in individuals with cancer.
4. Ongoing clinical activities
Early evidence suggests that combining checkpoint inhibitors with DNMTi is effective in a range of cancers including Hodgkin lymphoma, mammary carcinoma, mesothelioma, lung carcinoma and melanoma (Falchi et al., 2016).
Currently, only DNMT and HDAC inhibitors are being clinically investigated in combination with immunotherapy.

Table 1. Clinical trials combining epigenetic modulators and immune checkpoint inhibitors in cancer (Gallagher et al., 2017).
5. Challenges and promises
Regardless of which epigenetic modulator is explored in (pre)clinical studies, the toxicity on different tumor histologies remains challenging. A better understanding of the molecular mechanisms by which epigenetic inhibitors affect immune cells is therefore needed to improve their clinical development. Much also remains to be learned whether these epigenetic alterations are drivers of disease or consequences of the diseased state.
The epigenetic regulators referred to above are only a small fraction of the factors known to shape the chromatin landscape. A lot of epigenetic regulators remain relatively unexplored to date. Therefore, this field is at an exciting early stage and the diversity of writers, readers and erasers suggests an enormous unexplored potential.
The coming years should also provide an understanding of the safety, efficacy as well as possible acquired resistance of the most advanced epigenetic drugs as they enter the clinic which will allow further advances of the development of drugs that target new epigenetic players.
-----------------------------------------------------------------------------------------------------------
Authors:
Lijs Beke (Epigenetics professional) and Natasa Obermajer (I-O professional)



