
I-O Research: 2017 Recap & Proleptic Challenges for the Field in 2018
The unprecedented therapeutic benefits of immune-checkpoint blockers (ICB) have in the last two-three years been extended from the “traditional” immunogenic cancers, malignant melanoma and renal cell carcinoma (RCC) to other histologies classically described as “nonimmunogenic,” such as non–small cell lung cancer (NSCLC) (Topalian et al., 2012), and further in May 2017 - as a landmark event in oncology - led to the FDA approval of pembrolizumab (anti PD-1 antibody) for mismatch-repair (MMR)-deficient tumors (Figure 1). This represents a precedent of a drug being approved on the basis of a specific genetic profile rather than where the cancer originated.
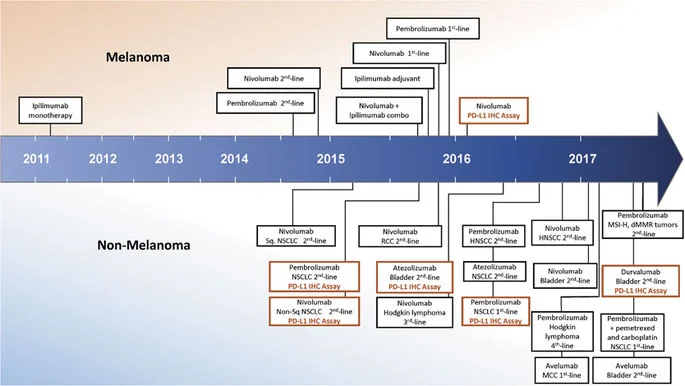
Figure 1. Timeline of FDA approvals for immune checkpoint blocking agents, including PD-L1 immunohistochemistry companion and complementary diagnostics (Taube et al., 2017).
The scientific basis behind this paradigm shift in anti-cancer treatment was published in Science. In their manuscipt Dr. Diaz (PI on the clinical trial) and his colleagues report in detail on results of the clinical trial [of 86 people in the trial with 12 different types of cancer more than half of the participants (46 patients) had an OR and 18 patients had a CR to pembrolizumab] and explain that the large number of neoantigens present in MMR-deficient tumors made them responsive to ICB therapy.
The limitations of above mentioned therapeutic successes of ICB have now gradually started to unveil the path forward and the next challenges the immuno-oncology field needs to face to expand treatments to a broader range of patients and increase the response rates:
1. Establish integrated, multidimensional biomarker to predict response to immunotherapy and drive patient selection.
ICB expression assessment by immunohistochemical (IHC) staining on tumor and/or tumor-infiltrating immune cells lacks sensitivity (some PD-L1-negative patients consistently experience clinical benefit), and specificity (not all PD-L1–positive tumors benefit from anti–PD-(L)1 therapy) (reviewed by Chabanon et al. (2015)), indicating the requirement for a companion biomarker(s) in predicting the response to ICB therapy.
(As stated above) Defects in the DNA repair machinery have now been consistently associated with improved survival and durable clinical benefit from ICB - MMR status predicted clinical benefit of anti-PD-1 blockage (Le et al., 2015 and 2017). These data rationalize screening of tumors for MMR/MSI [microsatellite instability (MSI) refers to the hypermutability of short repetitive sequences in the genome caused by impaired DNA MMR] for the selection of patients for immunotherapy. However, the frequency of MSI events is highly variable within and across tumour types (Cortes-Ciriano et al., 2017) - in analogy to highly variable tumor mutation burden across and within cancer subtypes (Alexandrov et al. 2013). It will therefore be important to understand the patterns of MMR across tumors and identify high yield tumor types (eg. gastric) or clinical scenarios (eg. progressive disease).
As for other mutational-based biomarkers that we should consider in predicting response in immuno-oncology, neoantigen burden has been shown to predict response to immunotherapy across tumor types. Carbone et al. (2017) published the results from the CheckMate-026 demonstrating that patients with both a high tumor-mutation burden and a PD-L1 expression level of ≥50% did experience a significant benefit from immunotherapy compared to those with only 1 of these factors or neither factor, further underscoring the impact of tumor mutation burden in immunotherapy response. McGranahan et al (2016) showed enhanced sensitivity to ICB in patients with advanced NSCLC and melanoma in tumors enriched for clonal neoantigens. Interestinlgy, their data suggest that in contrast to clonal neoantigens (attributed to smoking-induced mutations), therapy-induced (radiation or cytotoxic exposure) subclonal mutations fail to drive an efficient antitumor response. Goodman et al. (2017) corroborated that the tumor genomic landscape, mutational load, and tumor-specific neoantigens are potential determinants of the response to ICB and can influence patients' outcomes upon immunotherapy.
In December 2017 a noteworthy presentation on the topic by Drs. Rimm, Diaz and Sholl was published "The Present and the Future of Cancer Immunotherapy Biomarkers: Challenges, Opportunities, and Implications for Pathologists".
2. Understand the patterns of immune responsiveness and resistance to immunotherapy.
Enumerating the underlying mechanisms of de-novo (or primary) and acquired resistance to ICB has now become a logical next step for cancer research. While Sharma et al. (2017) comprehensively discussed known resistance mechanisms and provided rationale for combination therapies to overcome resistance [also briefly discussed below], Syn et al. (2017) reviewed the biological processes that operate in the formation of so-called immunoresistant niches and elucidate on the idea that converging mechanisms might underlie de-novo and acquired immunotherapy resistance.

Figure 2. Known Intrinsic Mechanisms of Resistance to Immunotherapy (A) Intrinsic factors that lead to primary or adaptive resistance including lack of antigenic mutations, loss of tumor antigen expression, loss of HLA expression, alterations in antigen processing machinery, alterations of several signaling pathways (signaling through MAPK and loss of PTEN expression, which enhances PI3K signaling, expression of WNT/b-catenin, loss of IFNg signaling pathways), and constitutive PD-L1 expression. (B) Intrinsic factors that are associated with acquired resistance of cancer, including loss of target antigen, HLA, and altered interferon signaling, as well as loss of T cell functionality.

Figure 3. Known Extrinsic Mechanisms of Resistance to immunotherapy. This includes CTLA-4, PD1, and other immune checkpoints, T cell exhaustion and phenotype change, immune suppressive cell populations (Tregs, MDSC, type II macrophages), and cytokine and metabolite release in the tumor microenvironment (CSF-1, tryptophan metabolites, TGF-b, adenosine).
As a succession on the new paradigm that differential activation of tumor-intrinsic Wnt/beta catenin pathway can explain the phenomenon of immune exclusion in a subset of cancers (Spranger et al., 2015) and further identification of specific oncogene pathways promoting the resistance to immunotherapy (Peng et al., 2016), studies that have used massively parallel sequencing shed light on the rich functional landscape of mutations, that through clonal evolution and selection [particularly acquired defects in interferon receptor signalling and antigen presentation] endow tumour cells with the ability to evade T-cell-mediated immunosurveillance.
At 32nd Annual Meeting of SITC Dr. Marincola presented a ‘theory of everything’ to try to explain patterns of immune responsiveness and resistance proposing that cancer cells go through a conserved evolutionary bottleneck, facing a ‘two option choice’ to evade recognition by the immune competent host: cancer cells either adopt a clean oncogenic process, devoid of immunogenic stimuli (immune silent tumors) or, alternatively, follow an entropic biology that tends towards immune recognition (immune active tumors) but resists rejection by recruiting compensatory immune suppressive processes. These different strategies represent two entirely different types of cancer, and strategies aimed at enhaning antitumor immunity.
3. Evaluate tumor immunogenicity.
Immunogenicity relies on a combination of antigenicity (provided by neo-epitopes) and adjuvanticity (conferred by specific damage-associated molecular patterns (DAMPs).
The very high attrition rate from a high mutational burden to the very few neoepitopes that will eventually produce an antitumor immune response illustrates the complexity of predicting tumor immunogenicity using genomic data alone. Namely, several other mechanisms apart from high mutational burden associating with neoantigen formation, such as oncogenic stress, secretion of immunosuppressive cytokines, or downregulation of MHC class I modulate tumor immunogenicity.
A step forward in understanding why some tumors are more aggressive than others presents mathematical modelling that can predict rationally which neoantigens will be the most effective at stimulating an immune response and how a cancer patient will benefit from certain immunotherapies (Luksza et al., 2017; Balachandran et al., 2017).
4. Define novel combinatorial approaches overcoming the resistance to ICB.
The strategies aimed at enhancing the effectiveness of immunotherapy will need to consider the importance of both, antigenicity and adjuvanicity, in anti-tumor immunity. With the above mentioned limitations of single predictive biomarkers, such as PD-L1 staining, multiple dimensions of data will be needed to capture the range of prognostic information, and provide a rational foundation to develop, select and combine IO agents in a way that inflames cold tumors, and coaxes tumors that are neither hot nor cold towards a more immunogenic phenotype.
4.1. Compared to tumor-associated antigens (TAA) that were used in the first
clinical trials either as peptide-based vaccines, part of the in vitro-generated and fully activated DCs or vaccines developed based on viral or bacterial vectors, neo-antigens are tumor-specific and immunogenic. Neoantigens are becoming the focus of intense research and may lead to a resurgence in cancer vaccine development. With the decreasing cost of next-generation sequencing, peptide manufacturing, and improvement of in silico prediction of peptide immunogenicity, the potential use of neoantigens is becoming feasible in anti-cancer treatment .
In the compelling webinar (recorded Oct. 2017) as part of the "Cancer Immunotherapy and You" webinar series, Dr. Robert D. Schreiber discusses the latest advances in technologies that identify unique genetic mutations in patients' tumors that make fitting targets for therapeutic intervention with cancer vaccines.
4.2. Apart from promoting the release of tumor neo-antigens, immunogenic cell death (ICD) stimulates immune adjuvant effects, functioning like an in situ vaccine (reviewed by Galluzzi et al. (2017). Immunogenic chemotherapy and radiation therapy elicit ICD Dr. Welsh presented the latest update on a phase II trial examining a combination of high-dose radiation therapy plus ipilimumab for patients with various types of stage IV cancers at the ASTRO’s 59th Annual Meeting, 2017, showing that radiation may help turn the tumor into a vaccine to stimulate an immune response.
In analogy, ICD and changed TME triggered by oncolytic virotherapy may explain improved efficacy of anti-PD-1 therapy in a phase 1b clinical trial testing the impact of oncolytic virotherapy with talimogene laherparepvec [first FDA-approved oncolytic virus for intratumoral therapy of nonresectable metastatic melanoma] in combination with the anti-PD-1 antibody pembrolizumab, published by Rivas et al. (2017). Promising antitumor activity was similarly demonstrated in a phase 2 randomized trial of talimogene laherparepvec combined with the ipilimumab alone (Chesney et al., 2017). The next generation of oncolytic viruses will likely combine multiple genetic modifications (transgenes and viral genetic alteration) that act to synergistically target tumors through multiple mechanisms.
4.3. Stimulating innate immunity is not a new concept in immunotherapy. The promising innate immune targets include CD40, Toll-like Receptors (TLRs, reviewed by Rokoff-Nahoum et al., 2009), RIG-I-like Receptors (RLRs), and Stimulator of Interferon Genes (STING, reviewed by Vargas et al., 2017). At the present, identifying strategies to optimize the triggering of multiple innate immune sensing pathways concurrently might be important to consider from the anti-tumor therapeutic perspective. For example, RIG-I was recently shown to be essential for radio/ chemotherapy sensitivity. On the other hand, STING dependent mechanism, which is also critical for spontanoues T cell priming depends on an alternative signaling pathway to ICD induced by some chemotherapeutic agents. While chemotherapy releases calreticulin (interacting with CD91), HMGB1 (a ligand for TLR4) and ATP (activating the inflammasome pathway to produce IL-1), STING acts as an adaptor protein linking DNA sensors to activation of IRF3 and NFkB and induction of type I IFNs.

Figure 4. Combination Immunological Interventions Can Form an Effective Combinatorial Cancer Immunotherapy. The 3pRNA-mediated MDSC reprogramming and reversal of the tumor’s immunosuppressive environment will ensure that high levels of IFN-a will be produced locally.MDSC reprogramming should be accompanied by a vaccination optimized to induce maximum CD8+ T cell immunity. A large panel of antigens covering CD4+ helper T cell and CD8+ cytotoxic T-cell-specific epitopes should be employed in the form of overlapping synthetic long peptides (SLPs). Coupling these SLPs directly to an adjuvant augments dendritic cell (DC) activation and subsequent antigen presentation. Choosing the adjuvant to ensure IL-12 and IFNa production by the activated DCs augments T cell priming and effector function. By using systemic antibody-mediated CTLA4 or PD-1 blockade, physiological immune checkpoints to limit T cell proliferation upon activation or T cell effector function upon target cell recognition, respectively, are blocked. T cells proliferate in the IL-12 and IFNa inflammatory environment, and tumor cell killing can initiate upon target cell recognition. (Boorn et al., 2016)
While immunostimulation with TLR agonists in cancer therapy has been investigated in several clinical trials (reviewed by Iribarren et al., 2016), the ongoing phase I study to evaluate STING pathway activator ADU-S100 as a monotherapy in patients with cutaneously accessible metastatic solid tumors or lymphomas was initiated in May 2016 and being followed up in September 2017 with phase Ib trial of ADU-S100 in combination with CIB in patients with accessible solid tumors or lymphomas.
4.4. Apart from CTLA-4 and PD-L1/PD-1, other immune checkpoint molecules have crowded the I-O space in the last couple years. The 'hottest' potential targets are VISTA, BTLA, TIM-3, LAG-3, GITR, TIGIT. While these molecules dampen the immune system, the antitumor immune T cell response is activated through agonistic targets such as 4-1BB, ICOSL, CD70 and OX40. Although many of the new agents show efficacy in vitro that demonstrate their desired function, turning these in vitro successes into clinical benefits and discovering their appropriate combination to most effectively affect different signalling pathways remains challenging.
4.5. Another class of agents that will inevitably shape cancer immunotherapy in the next years are Epi-drugs. Current epigenetic therapies are primarily directed towards two functional categories of epigenetic regulators: DNA methyltransferase inhibitors (DNMTi) and histone deacetylase inhibitors (HDACis). Whilst their function in immune priming of cancer cells has been explored reasonably thoroughly, the
role of many other novel epigenetic drugs has yet to be established, including histone
methyltransferases inhibitors (eg. EZH2 methyltransferase inhibitor GSK126), bromodomain/BET inhibitors (eg. JQ1), and histone demethylase inhibitors (eg. INCB059872). Roughly 35 phase I and/or II clinical trials combining checkpoint inhibitors and epigenetic drugs in various cancer types are (about to) recruiting patients (Dunn and Rao, 2017).
5. Scientific progress in emerging novel disciplines
The onco-immunology field is still waiting for scientific advances in identification of a druggable tumor-specific targeting approach for metabolic reprograming of exhausted anti-tumor T cells. Restifo's group discusses potential strategies to modulate metabolism for improving the immune response to tumors (Kishton et al., 2017). Another compelling area is targeting immune cell plasticity. Recent data demonstrating that Th17 cells can convert into immunosuppressive regulatory T (Treg) cells in tumor microenvironment help reconcile the limited efficacy of Treg cells-depleting approaches (Downs-Canner et al., 2017). Microbiome is additional field of interest for cancer immunology. A step beyond previous studies that relied mainly on mouse models of cancer present two recent publications in Science. The scientists found that those who had the most diverse gut microbes were most likely to respond to the immunotherapy and tumour growth was reduced in mice that received faecal transplants from people who responded to immunotherapy (Gopalakrishnan et al., 2017). A second study showed that people who received antibiotics to treat infections shortly before or after starting immunotherapy did not respond as well to PD-1-blocking therapies. The presence of the bacterium Akkermansia muciniphila in both humans and mice was linked to better responses to immunotherapy (Routy et al., 2017). What we would like to understand now is what the mechanism of action behind these data is.
6. Managing side-effects of CAR T cell therapy
One of the most rapidly emerging immunotherapy approaches in 2017 has been adoptive cell transfer (ACT)(Figure 4, Johnson and June, 2017), of which the one that has advanced the furthest in clinical development is CAR T-cell therapy. The remarkable responses CAR-T cell therapy produced in small clinical trials in patients with advanced blood cancers resulted in 2017 in two CAR T-cell therapies being approved by the FDA, one for the treatment of children with acute lymphoblastic leukemia (ALL) and the other for adults with advanced lymphomas.

Figure 4. Side-by-side comparison of attributes of TCR and CAR gene-engineered T cells for targeting tumor antigens
Challenges and opportunities of CAR T cell therapy discusses Dr. Carl June in the WALS lecture (Sept. 2016).
Dr. Rosenberg's lab was the first to report successful cancer treatment with CAR T cells. Since, the 2nd-4th generation of CAR T cell therapy have now been significanly improved in the efficacy by integrating one (second) or more (third) co-stimulation signals and/or cytokines (fourth). From here the idea is to incorporate gene-editing technologies to CAR T cells and thereby further enhance their potency: while genome-editing tool CRISPR-Cas9 has been employed to introduce a CAR gene at a specific location in the genome (Eyquem et al., 2017), additional applicability for gene editing could be the deletion of immune checkpoint genes.
Nevertheless, in many aspects, it is still early days for CAR T cells and other forms of ACT. The most acute challenge for this approach present systemic side-effects: apart from "on-target, off-tumor effects", cytokine release syndrome (CRS) and cerebral oedema, along with other neurologic toxicities, that can lead to serious harm or even death. While early intervention with pre-emptive Tocilizumab decreases the rate of CRS, further investigation into the cause, prevention and treatment of the cerebral toxicities is needed.
One approach is simply Forecasting Cytokine Storms with New Predictive Biomarkers. The field is now moving in the direction of “suicide switch” CaspaCIDe (and the variations of this theme), which can be engineered into T cells to address the off-tumor toxicity. A key part of achieving this goal, however, will be improving CAR specificity for target cells - “on-target, off-tumor effects” can be fatal, particularly in solid tumors - the question remains whether CAR T cells will ever be effective against solid tumors. The specificity can be improved with bispecific CARs or by exploiting the hypoxic tumor microenvironment (Juillerat et al., 2017).
Additional emerging challenge present the long-term effects of CD19-CAR-T cells- demonstrating antigen loss is a frequent cause of resistance to CD19-targeted immunotherapy, and also partial or complete lineage switch in ALL as a consistent mechanism of CAR resistance (Jacoby et al., 2016). In a phase 1 trial testing a new CD22-CAR, Fry et al. (2017) recently demonstrated the clinical activity of a CD22-CAR in B-ALL, including leukemia resistant to anti-CD19 immunotherapy.
The lessons learned from heamatological malignancies will drive the science of CAR-T cell immunotherapies in solid tumors. The first and most critical challenge will be to determine a safe target - a protein absolutely tumor-specific, or only shared with non-essential tissues, instead of the currently-targeted TAA.

Figure 5. Coggle diagram of completed and ongoing TCR and CAR gene-engineered T-cell immunotherapy clinical trials (per ClinicalTrials.gov).
7. Pre-clinical models of efficacy and tolerability.
The clinical development faces its own challenges:
- the difficulty of extrapolating data from preclinical models (Twyman-Saint Victor et al., 2015): a striking example is the disappointing results of anti-CTLA-4 therapy in the B16 preclinical melanoma model and the subsequent efficacy in clinical trials of patients with melanoma; the reverse was shown in pancreatic cancer, when encouraging laboratory results were not borne out in the clinic. [read the next blog for insights on addressing this issue]
- determining the most appropriate techniques to assess toxicity: standard dose-limiting toxicity observation periods are usually too short to account for toxicity from immune checkpoint-blocking antibodies, which may take many weeks to set in [7]. Further to this, standard oncology drugs are developed based on the maximum tolerated dose (MTD) principle, in which dose is titrated until tolerability limits further escalation. However, in the realm of immunotherapy it may be preferable to dose to optimal immunologic effect, in order to avoid levels of immune stimulation that could adversely affect antitumor immunity through, for example, T cell activation-induced cell death.
- developing across-the-board dosing recommendations due to the differences in drug class-based dose dependency [eg. higher doses of ipilimumab are associated with improved survival and higher toxicity whereas dose-dependent effects are not typical with anti-PD-1 agents].
- the evaluation of response in early clinical studies [standard RECIST criteria compared to recently developed immune-related response criteria (irRECIST)].
8. Overcoming the obstacles to advancement of the most effective clinical trials.
There is a clear lack of coordination in PD-1/L1 combination trial landscape and fragmentation due to a large number of IO drugs in clinical development with significant duplication. The data underscore the need for more collaborations, a greater emphasis on multi-site studies (eg. Woodcock et al., 2017) with a more careful use of umbrella trial platforms to find faster and more efficient means of putting I/O agents through human studies.
.........................................................................................................................................................


